
The FDA issued its draft of the Computer Software Assurance (CSA) Draft Guidance for comments on September 13, 2022. Industry professionals are invited to provide comments thru November 14, 2022.
USDM has been studying, training, and implementing this risk-based CSA methodology for our customers for a few years. The FAQ below curates many questions from our customers and attendees to our various webinars with the FDA.
CSV → CSA at a glance
- From documentation to thinking: CSA emphasizes critical thinking and risk-based testing over volume-based documentation.
- Leverage vendor evidence: SaaS providers do much of the heavy lifting — CSA encourages reuse, CSV often duplicated it.
- Same bar, faster path: CSA isn't lighter compliance — it's the same regulatory bar reached more efficiently.
- Voluntary transition: FDA encourages CSA but does not mandate it — choose the right model per system.
Watch our recent webinar with the FDA’s Francisco Vicenty, Case for Quality Program Manager.
If you have questions that are not answered in this Q&A, please contact us at usdm@usdm.com.
What is the difference between computer system validation (CSV) and computer software assurance (CSA)?
If you think of the 80/20 rule, the current CSV methodology has manufacturers spending 80% of their time documenting and only 20% of their time testing. The FDA wants to flip this so that 80% of a manufacturer's time is spent on critical thinking and applying the right level of testing to higher-risk activities, while only 20% of their time is spent documenting (CSA methodology). This critical thinking should be focused on three questions:
- Does this software impact patient safety?
- Does this software impact product quality?
- Does this software impact system integrity?
Using a risk-based approach is nothing new, and organizations such as the International Society for Pharmaceutical Engineering (ISPE), author of the Good Automated Manufacturing Practice (GAMP®) have been advocating this for two decades.
CSA is a framework designed to help manufacturers achieve CSV. CSA will provide clarity on the stance and methodology used to determine what is high risk and what is not, therefore minimizing misinterpretation by manufacturers. The clarification in the CSA approach flips the paradigm to focus on critical thinking (risk-based), assurance needs, testing activities, and documentation – in that order.
Why is the FDA making this change?
Too much work is done for fear of regulatory punishment instead of fear of putting a poor-quality product on the market. For software not used in a product, manufacturers are referring to burdensome guidance that is more than 20 years old, trying to avoid FDA Form 483 observations and warning letters from FDA investigations and third-party consultants. Nothing should be done for fear of regulatory observations. Instead, the focus should be on testing for higher confidence in system performance and applying the right risk-based assurance rigor for a given level of risk to patient safety and product quality. The new CSA framework also enables manufacturers to "take credit" for prior assurance activity and upstream and downstream risk controls like vendor qualifications.
What does “software not used in a product” (or non-product software) mean?
Non-product software is any software that is not directly used in a medical device, Software as a medical device (SaMD), medical device as a service (MDaaS), or end-product. It includes all of the software used in manufacturing, operations, and quality system activities that would follow the 21 CFR Part 820.70(i) guidance and the General Principles of Software Validation for relative Predicate Rules.
Is this just for medical device companies? Does CDER endorse it for pharmaceutical companies?
The short answer is no, the new CSA framework isn't just for medical device companies. There are a lot of potential applications for all of the life sciences.
The FDA’s Center for Devices and Radiological Health (CDRH) is working on this new draft guidance in cooperation with the Center for Biologics Evaluation and Research (CBER) and the Center for Drug Evaluation and Research (CDER). It is founded on a true, risk-based approach that should be considered when deploying non-product, manufacturing, operations, and quality system software solutions such as:
- Quality management systems (QMS)
- Enterprise resource planning (ERP)
- Laboratory information management systems (LIMS)
- Learning management systems (LMS)
- Electronic document management systems (eDMS)
See Francisco Vicenty's response to this question in our recent webinar here:
Watch the complete webinar here.
Does CSA apply to software in a medical device (SiMD) or software as a medical device (SaMD)?
CSA intends to provide recommendations to life sciences companies on computer software assurance for computer systems and automated data processing systems that are part of medical device production or the quality system. It does not apply to the design verification or validation requirements for software in a medical device (SiMD) or software as a medical device (SaMD). It also does not apply to technology vendors following a software development lifecycle (SDLC) that uses a risk-based approach for functional testing. Technology vendors are not regulated by the FDA.
What is an indirect system versus a direct system?
Indirect systems do not directly impact patient safety or product quality (for example, tools used in your CSV process like bug tracking systems or load testing and lifecycle management tools that do not directly impact the product). Indirect systems require less documentation.
Examples of indirect systems:
- Complaint handling systems
- Document management systems
- Bug tracking tools
- Testing tools
- Code control tools
Direct systems directly impact patient safety or product quality—like electronic device history or adverse event reporting—and may require increased testing based on risk. In other words, the riskier a system impact is on the end product, and the patient's safety, the more testing, and documentation are required.
Examples of direct systems:
- Automated calibration systems
- Electronic device history record
- Automated inspection system with no human checks
- Labeling systems
How do I evaluate risk in my application?
The FDA recommends using a risk-based analysis to determine appropriate assurance activities. Broadly, this risk-based approach entails systematically identifying reasonably foreseeable software failures, determining whether such a failure poses a high process risk to patient safety, product quality, or data integrity, and systematically selecting and performing assurance activities commensurate with the medical device or process risk, as applicable.
Those assurance activities can range from low-level risks, leveraging vendor testing activities, or ad-hoc testing may be sufficient to show the system meets its intended use. For higher-level risks, the system may require unscripted, ad-hoc, or scripted testing or a combination of the testing activities. There is no requirement to use any one specific risk evaluation method.
The graphic below shows one example of how you might evaluate the risk, but your evaluation methodology is ultimately up to your organization. USDM can help you think through this. Contact us to discuss your approach.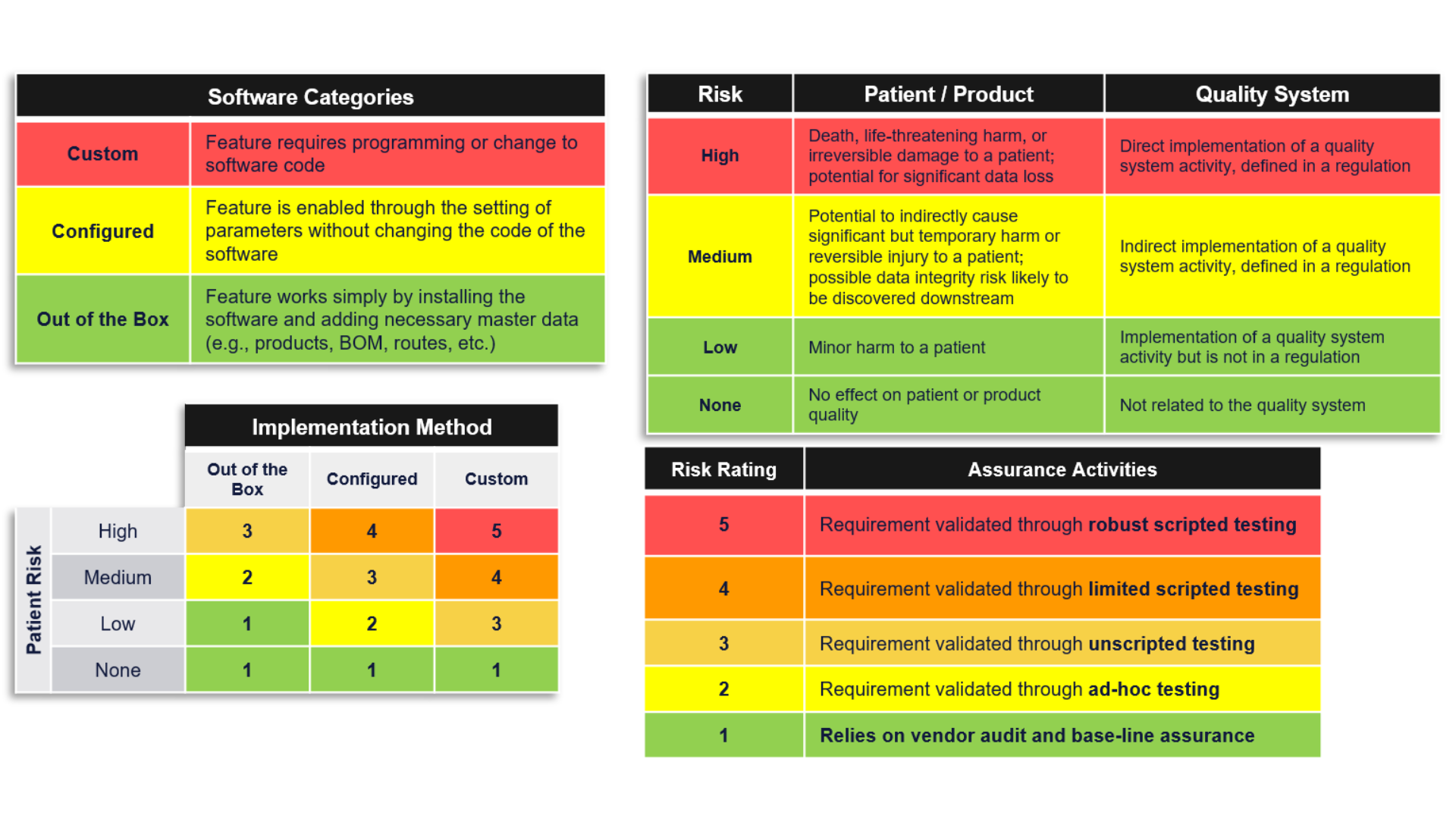
Can you provide examples of Ad-Hoc testing and unscripted testing? How do you show that you have done this type of testing?
Using the FDA framework unscripted testing can take two forms, ad-hoc and error-guessing. Ad-hoc testing generally consists of happy path testing while error guessing includes testing of failure modes as appropriate. In either case, a tester simply documents the following items as they perform their testing activities:
- Description of feature or function and failure modes that were tested
- Details around any failures that occurred during testing, including resolution
- Conclusion statement
- Record of who performed the testing and when
Additional information is required which could be included in other documents or part of the unscripted testing form:
- Intended use
- Risk determination
- Reviews and approvals as appropriate
- Supplier qualification
USDM has many SOPs, templates, and examples available for our customers. Please contact us to learn more.
The FDA has started citing companies for inadequate CSV efforts. How will inspectors be trained on the CSA initiative?
The FDA is undergoing an extensive training program for its auditors and is rolling out an agency-wide Case for Quality program. Further, the FDA is creating a Digital Center of Excellence, which will encourage manufacturers to reach out to the FDA to ask questions about their processes and procedures before an audit takes place. The goal is to provide more collaboration throughout the process and minimize this fear of regulatory observations that have led to misinterpretation of the original intent of the guidance.
Watch our on-demand webinar – Computer Software Assurance (CSA) Draft Guidance Webinar with the FDA – to see the FDA respond to this question.
Has the FDA reached out to other regulatory agencies such as MHRA, EU, etc., to verify that this approach is acceptable for companies that sell overseas?
Yes, the FDA has been working on the Case for Quality program in collaboration with its sister agencies abroad.
When does the FDA anticipate releasing this guidance?
The draft guidance was issued for comment in September 2022. Industry professionals are invited to provide comments thru November 14, 2022.
How can USDM help my company today?
The biggest hurdle to adopting CSA is the mindset and cultural behaviors your organization has come to believe. Four out of five senior leaders cannot explain WHY they do things the way they do them. Most respond, "we've always done it that way." Unless you can trace it to a law or a regulation – It's likely a tradition or legacy of misguided behavior holding you back. Changing internal perceptions and behaviors is the hardest part of evolving your validation processes. Using a 3rd party like USDM can help your organization stop inflicting processes and procedures that aren't required. USDM is on the cutting edge of technology and compliance, and we are closely watching the FDA's CSA guidance. We have progressive solutions and can save you significant time and money on your validation programs. Programs include:
- CSA Education and Training – USDM can help your team with a pilot project; train and mentor your teams on how to apply critical thinking; develop a risk-based approach, and consult on automated testing processes. We also offer organization change management programs.
- CSV/CSA Assessments – USDM will take a holistic approach to assess your current CSV process and make recommendations to get you to a true, risk-based CSA process according to your current state (i.e., quality of documentation, testing, SOPs/WIs, use of automation, and audit performance).
- CSV/CSA Methodology – USDM can revamp your entire CSV process and digitally transform it into a CSA process. From methodology development through end-user training, USDM will ensure your systems are compliant.
- Cloud Assurance – USDM provides a subscription service to deliver end-to-end GxP compliance of your cloud systems. USDM can manage and lighten your cloud validation burden from implementation through ongoing validation maintenance, including new releases.
How does the FDA define critical thinking?
As the manufacturer—the company and people producing the product—you know the business and the processes. You've got the best insight into how risk is introduced, where it matters, and what's going on from a process standpoint. Critical thinking is considering where the system could introduce a risk versus what is a product or process risk. This helps you tell your story, whether it is to the FDA or an arbitrary regulator and auditor. Demonstrate that you can tell that story and have the element of understanding and control about your product and your processes. There is no one-size-fits-all for any company or system. The FDA wants to know that you understand your processes and systems and are in control. Ensure that you can tell the FDA you know where the risk is being introduced, how you will mitigate the risk, and whether the controls you put in place are working.
What can activities or documents can I leverage from my technology vendor?
Whenever possible we recommend you verify a vendor's software development activities, including verification and validation, release management, change control, etc. We recognize that not all suppliers are willing to share this information or provide a mechanism to audit them. In those cases, you can rely on published vendor white papers, the vendor’s years of experience in life sciences and health care, their reputation, bug lists, etc., to evaluate the supplier and leverage their activities. USDM also conducts annual audits for many best-of-breed technology vendors through our Cloud Assurance service that can be leveraged as objective evidence. We also sell SOP templates, and vendor audits as needed.
What is the FDA doing to hold the software vendors more accountable to standardize their testing documentation so that it may be more readily and reliably leveraged by regulated life sciences companies?
FDA does not regulate software or cloud vendors. It is up to drug and device manufacturers to ensure the vendor has adequate controls in place or perform additional testing activities to ensure fit for use. Because FDA does not regulate these vendors, supplier qualifications should not include requirements for a complete quality management system. They merely need to have controls in place to ensure the output of quality applications for your intended use. USDM has created a Cloud Assurance Certification program that recognizes technology vendors that meet the quality and compliance demands of the life sciences industry and is THE badge of trust for GxP functionality. If you are exploring new technology vendors, we encourage you to consider Cloud Assurance Certified technologies as they have passed the rigorous compliance, security, and data integrity assessment by USDM and demonstrate their compliance with the consolidated global health authority statutory and regulatory requirements. We also make the Vendor Assurance Report available for any Cloud Assurance Certified technologies that can be leveraged as FDA evidence.
Does the FDA provide CSA certification for individuals or how can I get certified in CSA best practices?
At this time, there is no certification program specific to the CSA methodology, although there are multiple training courses available for Computer System Validation. CSA is simply a more effective and efficient way to ensure that your system meets its intended use when applied correctly. USDM offers training on the CSA methodology as well as practical, hands-on workshops to assist regulated organizations with applying this approach.
What does CSA mean for GAMP? Will GAMP become obsolete? Will it incorporate GAMP 5 second edition?
The impending CSA guidance is not going to create new concepts per se. It intends to simplify and clarify the use of non-product software and maximize testing efforts while minimizing documentation for lower-risk, non-product software systems. It is aligned with GAMP 5 and will be updated in future editions. CSA is what the FDA intended all along but lacked clarity, and the misinterpretation resulted in too much documentation for documentation's sake instead of better quality.
See Francisco Vicenty's response to this question in our recent webinar here:
Watch the complete webinar here.
What is the impact of 21 CFR Part 11?
CSA principles are applicable to 21 CFR Part 11, but the scope is narrowly focused. The primary concern is around system risk, intended use, and ensuring that you have confidence in the system.
What about audit trails?
Part 11, audit trail, is just a set of requirements and you must understand the best way to exercise those requirements. Know when you need more robust testing of those requirements and when you can just make sure that your vendor built that in. Overall, audit trails are not something that you need to expend a lot of extra resources and energy on.
What about ISO 13485?
ISO 13485 is integrated and well-written to incorporate risk-based thinking throughout all processes and applications. Nothing changes as it is based on a true risk-based approach.
What about MDSAP?
The FDA will make sure the medical device single audit program (MDSAP) is aligned with CSA down the road.
What does this mean for installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ)? What about User Acceptance Testing (UAT)?
The goal of CSA is to focus on critical thinking, do more business testing of the process and its intended use, and do less functionality testing.
Installation qualification: The vendor often does a good job of installation testing; still, it's smart to turn on the equipment, Login, and ensure it works. That's pretty low risk because it will be obvious if it doesn't work. Additional tasks would be ensuring you have all your required user manuals, vendor qualifications, and the like.
User acceptance testing (UAT): Focus on your business processes, how they work within the system, and how you want them to work within the system. This is where we expect to see much more testing and far less out-of-box functionality testing.
USDM Can Help
If you have questions that are not answered here or would like a consultation on your current CSV processes, please contact us at usdm@usdm.com.
Additional CSA References
- On-Demand Webinar with the FDA: Computer Software Assurance (CSA) Draft Guidance
- White Paper: Computer Software Assurance (CSA) Guidance. Here’s what you need to know
For practical transition guidance, see USDM's related post on how to align your CSV program with the FDA's CSA approach.
FAQ: CSV vs CSA Paradigm Shift
What is the difference between CSV and CSA?
CSV (Computer System Validation) emphasizes comprehensive documentation and broad testing. CSA (Computer Software Assurance) emphasizes risk-based testing and critical thinking, with greater reliance on vendor evidence and reduced documentation overhead.
Is CSA required by FDA?
No. CSA is encouraged through FDA guidance but not mandated. Organizations can continue CSV approaches, adopt CSA, or use a hybrid model based on system risk and organizational maturity.
Does CSA mean less rigorous validation?
No. CSA reaches the same regulatory bar through more efficient methods. The framework actually raises expectations for risk assessment and engineering judgment while reducing low-value documentation.
How does USDM help with the transition?
USDM provides CSV-to-CSA transition programs, validation strategy, training, and implementation services. Contact USDM.